For decades, the biopharmaceutical landscape has been dominated by Immunoglobulin G (IgG). Its favorable half-life, well-understood structure, and established manufacturing processes have made it the gold standard for monoclonal antibody (mAb) therapeutics. However, as the field of precision medicine advances and targets become more complex, researchers are increasingly looking beyond the traditional IgG framework. Non-IgG antibodies—specifically IgA, IgM, and IgE—are emerging as powerful new tools, offering unique mechanisms of action, superior avidity, and specialized tissue distribution that IgGs simply cannot match.
However, unlocking the full clinical potential of these alternative isotypes requires sophisticated antibody engineering. Unlike the relatively straightforward structure of IgG, non-IgG isotypes present intricate architectural challenges. They often feature highly sensitive hinge regions, heavily glycosylated Fc domains, and complex multimeric structures that demand precise control during development.
In this comprehensive playbook, we will dive deep into the critical aspects of non-IgG development, focusing on how engineering the Fc/hinge regions and controlling polymerization can overcome structural bottlenecks and yield highly efficacious therapeutic candidates.
The Paradigm Shift: Why Look Beyond IgG?
Before exploring the engineering strategies, it is essential to understand why non-IgG isotypes are worth the developmental effort. Each class brings distinct biological advantages to the table:
- IgA (Immunoglobulin A): As the primary antibody of mucosal immunity, IgA is uniquely positioned to neutralize pathogens at the exact sites of entry, such as the respiratory and gastrointestinal tracts. Furthermore, its interaction with the FcαRI receptor strongly triggers neutrophil-mediated antibody-dependent cellular cytotoxicity (ADCC), making it a highly attractive candidate for solid tumor oncology.
- IgM (Immunoglobulin M): IgM is the immune system’s first responder. Its multimeric structure (forming pentamers or hexamers) gives it unparalleled avidity. This makes IgM incredibly effective at targeting repetitive antigens, clearing viruses, and targeting tumor-associated carbohydrate antigens with low intrinsic affinity but high overall binding strength.
- IgE (Immunoglobulin E): Traditionally known for its role in allergies and parasitic defenses, IgE binds to its receptor (FcεRI) with an affinity orders of magnitude higher than IgG binds to its receptors. This extremely tight binding is now being harnessed in “AllergoOncology” to direct powerful macrophage and eosinophil responses against cancer cells.
Despite these advantages, natural non-IgG molecules often suffer from short serum half-lives, manufacturing instability, and rapid proteolysis. Overcoming these hurdles is where targeted antibody engineering becomes indispensable.
Mastering the Hinge Region in Antibody Engineering
The hinge region of an antibody is the flexible amino acid stretch that connects the antigen-binding Fab arms to the effector-driving Fc domain. It dictates the spatial flexibility of the antibody, determining how well the two Fab arms can stretch to bind simultaneously to target antigens. In non-IgG antibody engineering, manipulating the hinge is a delicate balancing act.
Flexibility vs. Stability
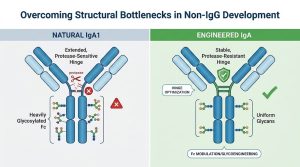
Take IgA as an example. Human IgA exists in two subclasses: IgA1 and IgA2. IgA1 features an extended, proline-rich hinge region. This provides exceptional flexibility, allowing the Fab arms to reach widely spaced antigens. However, this extended hinge is a double-edged sword: it is highly susceptible to cleavage by bacterial proteases, which can quickly neutralize the antibody’s effectiveness in mucosal environments.
Conversely, IgA2 has a truncated hinge, making it far more resistant to proteolytic cleavage but at the cost of reduced reach and flexibility.
Engineering Solutions for the Hinge
Through targeted antibody engineering, developers can create chimeric or humanized non-IgG molecules that capture the best of both worlds. Strategies include:
- Residue Substitution: Mutating specific proline or threonine residues within the extended IgA1 hinge to eliminate protease cleavage sites while retaining spatial flexibility.
- Hinge Swapping: Grafting the hinge region of a more stable isotype onto a non-IgG backbone to improve overall stability during bioprocessing without completely abolishing avidity.
- Disulfide Bond Engineering: Introducing novel cysteine residues near the hinge to form stabilizing disulfide bonds, locking the antibody into a highly stable conformation that extends its functional half-life in vivo.
Optimizing the Fc Domain for Enhanced Effector Function
The Fc (Fragment crystallizable) domain is the engine room of the antibody’s effector functions. It is the site that interacts with immune cells via Fc receptors and activates the complement cascade. In the context of non-IgG isotypes, the Fc/hinge interplay is highly complex and heavily influenced by unique glycosylation patterns.
Modulating Immune Receptor Binding
The primary goal of Fc engineering in non-IgG development is often to fine-tune the engagement with specific innate immune receptors.
- For IgA, engineering the Fc domain focuses on optimizing its affinity for the FcαRI (CD89) receptor found on neutrophils and macrophages, driving potent tumor cell killing.
- For IgE, researchers engineer the Fc region to exploit its ultra-high affinity for FcεRI on mast cells and basophils, aiming to direct a massive, localized immune attack against solid tumors while engineering safety switches to prevent systemic anaphylaxis.
Glycoengineering the Fc Region
Unlike IgG, non-IgG antibodies are heavily glycosylated. IgM and IgE, for instance, contain numerous N-linked glycosylation sites that are critical for their folding, secretion, and structural integrity. However, heterogeneous glycosylation during large-scale manufacturing can lead to batch-to-batch variability and altered pharmacokinetics.
Advanced antibody engineering techniques are used to delete non-essential glycosylation sites or to engineer cell lines that produce uniform, human-like glycan structures. This not only improves the homogeneity of the therapeutic product but can also be tailored to prevent rapid clearance by hepatic receptors, thereby extending the in vivo half-life of the non-IgG therapeutic.
The Complexity of Polymerization Control
Perhaps the most unique and challenging aspect of working with non-IgGs—specifically IgM and secretory IgA—is their polymeric nature. Unlike monomeric IgG, these isotypes assemble into large, multi-subunit complexes.
The Role of the Tailpiece and J Chain
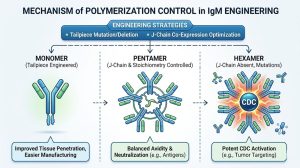
The polymerization of IgM and IgA is driven by an 18-amino acid C-terminal extension known as the secretory tailpiece. This tailpiece contains a critical cysteine residue (Cys575 in human IgM) that forms intermolecular disulfide bonds.
In vivo, IgM naturally forms pentamers (five monomers) in the presence of the Joining (J) chain, a small polypeptide that incorporates into the polymer and regulates its final structure. In the absence of the J chain, IgM can form hexamers (six monomers).
Engineering the Mechanism of IgM Polymerization
For therapeutic applications, controlling this polymerization process is paramount. Hexameric IgM, lacking the J chain, activates the complement cascade much more efficiently than pentameric IgM. Depending on the therapeutic goal—whether it is inducing massive complement-dependent cytotoxicity (CDC) against a tumor cell or avoiding systemic complement activation—engineers must precisely control the oligomeric state.
Engineering strategies to control polymerization include:
- Tailpiece Deletion or Mutation: By mutating the cysteine residues in the tailpiece, researchers can force IgM or IgA to express as stable monomers or dimers, which are easier to manufacture and have better tissue penetration than massive pentamers.
- J-Chain Co-Expression Optimization: To ensure a homogenous batch of purely pentameric IgM, manufacturing cell lines must be genetically engineered to co-express the heavy chain, light chain, and the J chain in exact stoichiometric ratios.
- Introducing Hexamerization Mutations: Conversely, if potent CDC is desired, specific point mutations can be introduced into the Fc domain that favor hexamer formation, resulting in a hyper-potent therapeutic candidate.
Managing the mechanism of IgM polymerization is not just a structural exercise; it is the definitive step in ensuring the therapeutic efficacy, manufacturability, and safety profile of multimeric antibody drugs.
Conclusion: The Future of Biotherapeutics is Diverse
The transition from a purely IgG-focused pipeline to a diversified portfolio including IgA, IgM, and IgE represents the next frontier in biologics. However, unlocking this potential requires specialized expertise in non-IgG antibody engineering. From stabilizing the highly flexible hinge regions and optimizing heavily glycosylated Fc domains, to mastering the complex biophysics of polymerization, success in this field demands innovative engineering solutions.
As developers continue to refine these structural elements, we will undoubtedly see a new wave of life-saving therapies that conquer the limitations of traditional monoclonal antibodies, offering hope for difficult-to-treat cancers, infectious diseases, and mucosal conditions.
Partner with Creative Biolabs for Your Engineering Needs
At Creative Biolabs, we specialize in the complex structural requirements of alternative antibody isotypes. We offer a comprehensive suite of customized engineering services to help you navigate the challenges of hinge optimization, Fc modulation, and polymerization control for your novel therapeutics.
Explore our specialized services below: