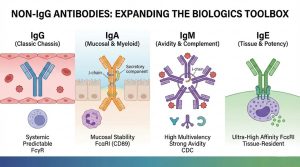
IgG has been the “default chassis” for therapeutic antibodies for decades—and for good reason: mature discovery workflows, predictable developability, well-characterized Fc effector biology, and scalable manufacturing. But the industry’s IgG-centric comfort zone has a downside: it can steer programs toward familiar formats even when biology suggests a different antibody class would be more fit-for-purpose.
That’s why non-IgG antibodies—especially IgA, IgM, and IgE—are re-emerging in modern biologics design. Not as a replacement for IgG, but as a strategic expansion of the antibody toolbox. As target biology becomes more nuanced (mucosal immunity, multivalent antigen clustering, myeloid engagement, tissue-resident effector cells, and microenvironment constraints), isotype choice is increasingly treated as an intentional design variable—not an afterthought.
Below is a practical, design-centric guide to what makes IgA/IgM/IgE different, where they can shine, and what you must engineer (and de-risk) to move from concept to a CMC-ready program.
Why Non-IgG Is Coming Back (And Why Now)
Three industry shifts are accelerating the non-IgG comeback:
1) Targets are getting harder—and more spatially complex.
We’re no longer only neutralizing soluble ligands or blocking a single receptor. Many programs now need to operate at interfaces: mucosal surfaces, tumor stroma, inflamed tissues, or immune synapses. Isotypes evolved for those contexts can offer mechanical advantages (transport, avidity, effector recruitment).
2) Fc biology is now a controllable design layer.
We’ve learned to tune Fc—effector function, half-life, receptor selectivity, glycosylation—and we now apply that thinking beyond IgG. Non-IgG Fc domains interact with distinct receptor families (e.g., FcαRI/CD89 for IgA; Fcε receptors for IgE), opening different effector “wiring.”
3) Engineering + analytics caught up with the pain points.
Historically, non-IgG formats were avoided due to stability, heterogeneity, and manufacturing challenges. Today, improved expression systems, better purification strategies, and deeper analytical toolkits are making these programs more executable—if designed carefully.
IgA Antibodies: Built for Mucosal Frontlines and Myeloid Engagement
What makes IgA fundamentally different?
IgA is the dominant antibody class at mucosal surfaces (respiratory, GI, urogenital). It can exist as monomeric IgA in serum and as dimeric/secretory IgA (sIgA) at mucosa, where it’s optimized for stability and function in harsh environments. That mucosal specialization is precisely why IgA is being revisited for biologics concepts tied to local immunity, barrier tissues, and luminal targets.
The Fc advantage: FcαRI (CD89) and neutrophil-centric effector options
A key design differentiator is IgA’s relationship with FcαRI (CD89) on myeloid cells, including neutrophils. This creates a pathway to strong myeloid effector recruitment that is mechanistically distinct from classic IgG–FcγR paradigms. In practice, IgA designs are often discussed in the context of neutrophil-driven cytotoxicity and phagocytic programs, which can be attractive for targets where myeloid cells dominate the local microenvironment.
A notable example of “why isotype matters” comes from preclinical/translation work in which an IgG anti-GD2 format was reformatted into an IgA1 isotype to alter tolerability and effector balance, while still enabling potent myeloid engagement.
Where IgA concepts tend to fit best
-
Mucosal pathogen interception and neutralization (think “block at the gate,” not only in blood)
-
Barrier-proximal oncology targets where luminal or surface-accessible antigens are relevant
-
Myeloid-biased effector strategies, especially when neutrophils/macrophages are critical actors
The practical trade-offs you must design around
IgA programs often succeed or fail on developability details:
-
Shorter serum half-life vs. IgG (often requiring Fc/format strategies to match dosing goals)
-
Complex glycosylation and heterogeneity (impacts stability, potency, and analytics)
-
Multiple formats (monomeric IgA vs dimeric/sIgA) that shift both biology and CMC complexity
Bottom line: IgA isn’t “IgG but different.” It’s a different biological instrument—powerful, but demanding disciplined engineering choices early.
IgM Antibodies: Multivalency, Avidity, and Complement Power—By Design
IgM’s structural superpower: avidity through multimerization
IgM typically assembles into pentamers (often stabilized by a J chain), and can also appear as hexamers. This architecture yields many antigen-binding sites, enabling exceptionally strong functional binding through avidity—particularly valuable for low-affinity interactions, repetitive epitopes, or antigens that benefit from clustering.
Why avidity matters in modern biologics
Avidity can rescue programs where:
-
The target density varies (heterogeneous expression across cells)
-
The epitope is weakly immunogenic or structurally constrained
-
You want to drive receptor clustering or enhance local retention
IgM’s multivalency also makes it interesting for agglutination-like mechanisms and for binding “difficult” antigen presentations that don’t behave well under monovalent/low-valency assumptions.
Complement activation is not an afterthought for IgM
IgM is widely recognized for efficient complement activation relative to many IgG contexts (with substantial nuance depending on format and biology). For certain concepts—especially those where CDC-like mechanisms are strategically desired—IgM can serve as a more naturally aligned chassis.
The engineering/CMC reality check for IgM
IgM is big. And big molecules demand more from the entire program:
-
Expression & assembly control (getting the “right” multimer distribution)
-
Purification and stability (aggregation risk, shear sensitivity, denaturation during processing)
-
Analytical characterization of size variants, J-chain incorporation, and glycoforms
In other words: IgM can deliver biology that IgG simply can’t replicate—but you must budget for CMC sophistication from day one.
IgE Antibodies: Tissue-Resident Effector Biology and Ultra-Potent Signaling—With Safety Front and Center
IgE biology is uniquely “amplified”
IgE binds with high affinity to Fcε receptors, classically associated with mast cells and basophils, and more broadly with immune activation pathways that can be extremely potent. That potency is exactly why IgE is simultaneously fascinating and approached cautiously.
Why IgE is being explored for oncology and immune activation concepts
The renewed interest is partly driven by the recognition that tissue-resident effector cells and microenvironment-specific immune wiring can matter as much as circulating effector mechanisms. Clinical exploration of IgE antibodies in cancer has reported a manageable safety profile and early signals consistent with the idea that IgE therapeutics behave differently from IgG drugs.
The non-negotiable: safety-by-design
For IgE, design is inseparable from risk management:
-
Epitope and target selection must avoid unintended crosslinking hazards
-
Functional assays must address Fcε biology explicitly (not “IgG-style” proxies)
-
Formulation and dosing logic often require additional safeguards and staged validation
IgE is not “the next IgG.” It’s closer to a high-gain system: promising, but only when the program is architected with rigorous control points.
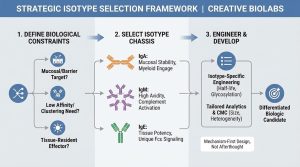
Choosing the Right Isotype: A Design Framework That Actually Works
Instead of asking “Which isotype is best?”, ask “Which constraints define success?” Here’s a practical framework used in advanced biologics design:
1) Where does the antibody need to work?
-
Bloodstream/systemic → IgG often wins on half-life and platform maturity
-
Mucosa/lumen/barrier → IgA (and secretory formats) become strategically relevant
-
Tissue-resident immune niches → IgE may unlock distinct effector options
2) What binding physics does the target demand?
-
Low affinity but high density → IgM avidity can be a decisive advantage
-
Need receptor clustering → multivalency (IgM) or tailored formats (IgA/IgG hybrids)
-
Need pure neutralization → any isotype can work, but Fc-driven biology changes the story
3) Which effector cells must you recruit?
-
Neutrophil/myeloid-biased mechanisms → IgA–FcαRI pathways can be compelling
-
Classic NK/macrophage FcγR engagement → IgG remains the standard
-
Fcε receptor–associated effector responses → IgE (with strict controls)
4) What does “developable” mean for this program?
Define acceptable ranges for:
-
Aggregation propensity, viscosity, size heterogeneity
-
Glycosylation control (especially for IgA/IgM)
-
Stability under process stress (filtration, mixing, freeze–thaw)
-
Analytical release panel feasibility
When teams evaluate non-IgG using IgG-only assumptions, they often misjudge risk. Non-IgG isotypes require isotype-appropriate developability criteria.
Common Pitfalls That Slow Non-IgG Programs (And How to Avoid Them)
Pitfall A: Treating isotype selection as a late-stage “format swap.”
If you’re serious about IgA/IgM/IgE, select the isotype early enough to build the right assays, analytics, and CMC strategy.
Pitfall B: Using IgG-era potency assays only.
Non-IgG mechanisms often require different readouts: receptor-specific activation, myeloid engagement assays, complement-focused profiling, mucosal stability simulation, and crosslinking risk evaluation.
Pitfall C: Underestimating heterogeneity.
Size variants (IgM), glycoforms (IgA/IgM), and functional receptor engagement differences can turn into late surprises. Build an analytical plan early.
Pitfall D: Ignoring “delivery context.”
IgA’s value is tightly linked to mucosal context; IgE’s value may be tightly linked to tissue-resident effector biology. If the biology doesn’t match the isotype’s evolutionary “job,” you’ll fight the format instead of leveraging it.
Where Non-IgG Biologics Are Most Strategically Useful Today
You’ll see the strongest rationale for IgA/IgM/IgE in programs that demand at least one of the following:
-
Mucosal interception, stability, or transport (IgA-focused concepts)
-
High-avidity binding to difficult antigens (IgM-focused concepts)
-
Distinct immune wiring and tissue effector leverage (IgE exploration with rigorous controls)
The shared theme: mechanism-first design. When mechanism and format align, non-IgG can be a genuine differentiator—not just a novelty.
How Creative Biolabs Supports Non-IgG Antibody R&D (Services)
If you’re planning an IgA/IgM/IgE program, Creative Biolabs provides end-to-end support across discovery, engineering, and CMC-enabling execution: